





DESCRIPCIÓN


La enfermedad afecta a ambos sexos por igual y se ha observado que es postraumática en numerosos casos. Esta enfermedad puede aparecer en cualquier momento de la vida, desde la infancia hasta la edad adulta. Suele manifestarse de forma unilateral e incluye proliferación linfática focal en un hueso o bien en huesos contiguos (como es el caso de las vértebras). El tejido blando adyacente también puede verse afectado, lo que produce atrofia muscular difusa. Puede resultar afectada cualquier parte del esqueleto. Cuando afecta a la caja torácica lo hace de forma progresiva y puede poner en peligro la vida del paciente. El proceso patológico es la sustitución de hueso normal por un tejido linfático que se expande de forma agresiva. Se desconoce cuál es el estímulo que origina este cambio en el hueso, y si todos los huesos del individuo afectado resultan igual de propensos a reaccionar de esta forma adversa. Debido a que se observa con mayor frecuencia en casos de traumatismos, resulta razonable deducir que el episodio traumático inicia la secuencia de acontecimientos y, debido a que la neovascularización del coágulo de sangre que se forma entre los fragmentos de una fractura es el primer paso de la curación de un hueso, puede especularse que la enfermedad de Gorham tiene su origen en la aparición de algún error en este proceso, o en el control de este proceso. El tejido neovascular, que presenta una proliferación agresiva, parece ser el agente causante de la pérdida ósea masiva que se produce en esta enfermedad. Viagra 100mg: Le médicament qui a fini par avoir un effet involontaire


PROBLEMAS
Ausencia de atipia celular.
Escasa respuesta osteoblástica/ausencia de ésta/ calcificación distrófica.
Resorción ósea progresiva local.
Lesión no ulcerosa y no expansiva. Kamagra oral jelly saved my sex life because it treats my ED issues without having to swallow pills, which I can't tolerate
Patrón radiográfico osteolítico.
Ausencia de una causa hereditaria, metabólica, neoplásica, inmunológica o infecciosa.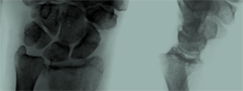
Este trastorno óseo se atribuye a una proliferación endotelial linfática focal, en la que el hueso es destruido y reabsorbido de forma progresiva. El curso esperado de acontecimientos después de una fractura de un hueso afectado por la enfermedad de Gorham es el fallo de la curación seguido de una reabsorción progresiva de hueso alrededor del lugar de la fractura. Se ha observado que la progresión de la enfermedad se detiene sin tratamiento en numerosos casos, si bien no se espera la regeneración del hueso destruido. El curso autolimitado de la enfermedad se correlaciona bien con dos fases de evolución de las lesiones histopatológicas con proliferación de tipo neoplásica de las células endoteliales linfáticas, que corresponden a la destrucción rápida y masiva del hueso, y una diferenciación posterior de las células en

Los criterios para diferenciar entre lo que es y lo que no es enfermedad de Gorham todavía no están bien desarrollados. El grupo más fácil de separar es el de la osteólisis multicéntrica, en la que los primeros afectados en la gran mayoría de los casos son los pies y las manos, la denominada acroosteólisis. Se ha descrito un subconjunto claramente hereditario y se han reconocido dos síndromes. Cabe destacar que existe otro grupo de pacientes con osteólisis multicéntrica que presenta una nefropatía asociada.
SOLUCIONES
Aparte del tratamiento ortopédico estándar para las fracturas, no uniones y deformidades, el tratamiento médico de la enfermedad de Gorham se divide en tres grupos:
2. Medicación antiosteoclástica (bifosfonatos) e interferón. La acción sinérgica del ácido zoledrónico y el interferón es un tratamiento antiangiogénico potente, que es el que actualmente está ofreciendo los mejores resultados.
3. Radioterapia: la radioterapia actúa acelerando la esclerosis de los vasos sanguíneos que presentan proliferación y evita que estos se regeneren. A pesar de que el uso de dosis totales de 30 a 45 Gy ha resultado efectivo, algunos autores obtuvieron excelentes resultados utilizando una dosis total de 15 Gy en un caso en el que estaba afectada la extremidad superior. Otros informaron de un alivio rápido de los síntomas y de la prevención de mayor destrucción ósea durante un período de seguimiento de 6 años con una dosis total de 3000 rads. La regeneración del hueso tras el tratamiento con radiación no es frecuente, pero se ha dado en algunos casos. Los niños presentan un riesgo significativo de desarrollar un sarcoma a largo plazo.
El pronóstico varía desde la discapacidad leve hasta la muerte, dependiendo de si se ven afectadas las estructuras esqueléticas vitales. Se ha notificado que más del 15% de los pacientes muere a consecuencia de su enfermedad. Los casos con afectación de la pelvis, el tórax y las vértebras cervicales producen discapacidad grave.
Las complicaciones neurológicas aumentan la mortalidad hasta un 33,3%. Sin embargo, normalmente la enfermedad permanece localizada en una región esquelética y se detiene de forma espontánea.
Se están comenzando a estudiar los mecanismos genéticos y ya se ha identificado un receptor de factor de crecimiento endotelial vascular (VEGFr3), el cual es específico del sistema linfático. Asimismo, se han identificado tres genes asociados al linfedema hereditario (inflamación generalizada debido a una anomalía linfática). En la actualidad, se están estudiando modelos animales que simulan el linfedema. Resulta posible que la patogénesis de la linfangiomatosis y de la enfermedad de Gorham sea diferente. Las malformaciones linfáticas que aparecen en la primera infancia pueden originarse a partir de la formación aberrante de vasos linfáticos, mientras que la proliferación adquirida de pequeños vasos linfáticos observada en la enfermedad de Gorham puede derivarse de factores de crecimiento linfangiogénicos. En particular, la IL-6 parece ser un mediador de la osteólisis masiva en pacientes con el síndrome de Gorham-Stout.
Deseamos destacar la necesidad de adaptar el tratamiento de acuerdo con la presencia de dianas terapéuticas y las complicaciones que estos tratamientos pueden inducir en un paciente.
2. Animamos a los pacientes y médicos a que, tan pronto como se sospeche el diagnóstico, se pongan en contacto con los centros con equipos multidisciplinares que participan en el tratamiento de la enfermedad con protocolos activos y con gran experiencia.
3. Tras el análisis de la manifestación clínica, los hallazgos en las imágenes y las muestras histológicas disponibles, deben clasificarse en uno de los 5 grupos siguientes dependiendo de las características del hueso y del tejido blando:
> Osteólisis multifocal sin anomalía linfática del tejido blando.
> Osteólisis focal con anomalía linfática del tejido blando.
> Osteólisis multifocal con anomalía linfática del tejido blando.
> Osteólisis linfática en el contexto de tumores vasculares o síndromes.
Dr Juan Carlos López-Gutiérrez
Centro de anomalías vasculares Hospital Infantil
La Paz
Madrid, España
